近年來由於生物檢測技術及網際網路的快速發展,以前需要花費大量時間與精力才能收集的生醫資料漸漸轉為可以快速累積的數據。這個情況使得過往需要透過大量閱讀文獻做出明確假設才能進行的生醫研究,逐漸由假設驅動(hypothesis-driven)的實驗方法,走向了另外一條可能的路線──無假設研究(hypothesis-free)。
無假設研究
無假設研究讓研究人員不需要在事前就預期會看到甚麼樣的資料變化,亦或僅能針對少量的分析標的進行追蹤,取而代之的方式則是透過全面性的資料收集,根據資料的變化情形來提出後續的研究假設,進而大幅減少初始假設錯誤的機率,也更能減少因既定的生物醫學知識限縮了假設的可能性。舉例來說,過往在針對臺灣非吸菸女性肺癌組織尋找具有顯著表現量變化的基因時就直接收集病患的病變組織進行全轉錄體(transcriptome)的研究,其結果意外地發現在癌變的組織上神經細胞攀爬相關的基因表現量具有明顯變異。然而,生理學告訴我們人類的肺臟是沒有神經的,若我們採用假設驅動的方法進行此項研究,不太可能把神經細胞相關的基因列為主要的研究標的,更遑論找到這些神經相關基因與肺癌之間的相關性。研究結果發現這些神經攀爬相關的基因表現量與女性肺癌病患的存活具有顯著關係,其可能的機制為這些基因雖然過往被標記為神經細胞攀爬,但也參與在細胞移動時組成與拆解細胞骨架,而這些與癌細胞的移動具有高度相關,也正好呼應了無假設研究上可能跳脫既有的知識框架找到新的研究標的。
無假設研究具有其資料優勢可以找到過往未曾被報導過的數據,但由於高通量的資料累積,如何有效率且正確的處理與分析資料成了亟待解決的問題。傳統的生醫研究能獲得數百筆數據已是相當不容易,但高通量的研究則在短時間內給予研究人員單一個體數萬筆以上的數據,近年來資料量更是大幅度成長到數百萬筆以上,此時生物資訊及生物統計在生物醫學類的健康數據上便極為重要。以基因研究為例,2000年時代的研究可能多為研究人員針對數個基因進行表現量研究,因此在資料分析上僅需執行數個基因的統計分析,利用手算亦或是商用套裝軟體即可達成此一目標。然而,隨著高通量基因體資料的出現,全轉錄體的研究會產生出數萬筆資料,此時透過程式進行相關分析已是不可或缺。
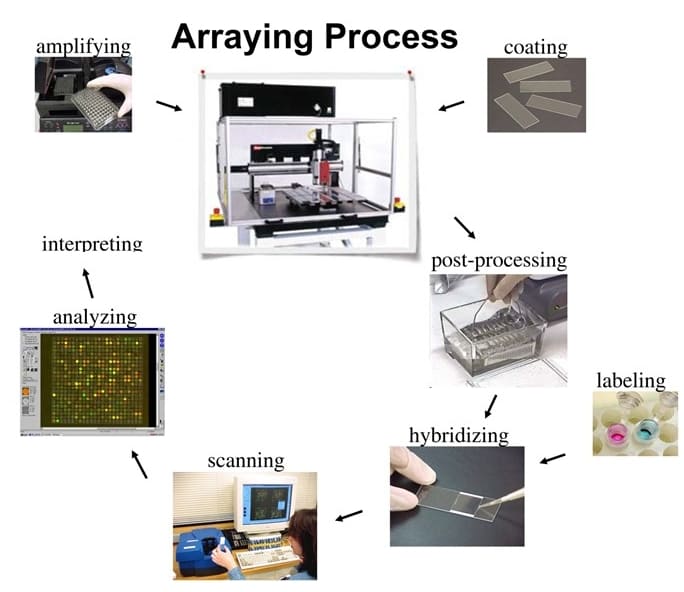
圖1:微陣列的製造過程。
此外,由於進行非常大量的統計檢定,如何在確保程式執行效率外,卻又不因檢定次數的增加而大幅提升偽陽性(false positive rate)就會是生物統計方法學上的重要角色。舉例來說,過往微陣列(microarray)基因表現量數據在肺癌的研究上找出了許多重要的生物標記(圖1),我們可以透過這些生物標記的表現量來預測病患的存活情形。然而在實務運用上卻遇到了很大的阻礙,其中最主要的困難點來自於不同的族群及資料集間的一致性相當的低,不同的資料集找尋出的生物標記皆不同,且彼此的交集比例近乎於零,由於我們無法預期新的病患是否會與既有的資料集相似,因此這個障礙讓實際運用此類生物標記的可能性降到極低。
為了解決這個問題,過去透過生物資訊的手法進行了功能性的基因群集研究,目標為找出跨資料集間共通的肺癌病患存活生物標記。考量不同資料集間的特性差異,若針對每一個基因進行檢定,高次數的統計檢定將讓誤差疊加而產生放大效果,我們決定利用基因的細胞功能將其群集化,如此可以從原先數萬筆的檢定次數大幅下降到數十次,再透過排列法(permutation)重複隨機抽取相同基因數目的虛無基因群(null hypothesis)去測試特定功能的基因群是否存在顯著預測病患存活的效果。本研究的分析結果證實藉由生物功能將基因群集化後進行分析,能夠增加找到跨資料集中具有共通性預測能力的基因群,最終我們找到了16個與細胞凋亡執行週期(apoptotic execution phase)相關的基因群可以用來預測肺癌病患的存活情形,也證實了藉由生物資訊方法將能夠增加分析的效率,並提升成果的一致性。
基因演算法
生物資訊方法除了作為學術上的研究之外,已經漸漸有相應的商業化基因檢測產品問世。舉例來說,目前乳癌病患可以透過癌症組織的基因表現量進行分型,且其基因表現量的分型結果不僅可協助預測病患的存活情形,更能作為未來治療決策的參考。想像下列的情境:當民眾被診斷得到癌症時,在害怕癌症可能復發或轉移的情形下,通常會選擇較為積極性的治療方法,藉由化學治療或是放射線治療來根除可能殘存的腫瘤細胞。然而,是否每個病患都需要接受較為積極性的治療方法存在著爭議,因此若能有基因檢測產品提供癌症病患的復發機率做為參考,將對病患的治療決策做出更好的選擇(圖2)。在此想法下,我們藉由收集一二期卵巢癌病患的癌症組織基因表現量,做為起始的訓練資料集。希望可以在訓練資料集上找到最適合用來預測病患復發的基因組合,然而,在這個目標上遇到的第一個障礙即為如何從5萬個以上的探針挑選出合適的基因組合。假定我們希望在5萬筆的基因探針中找到最適合的10個基因組合,則窮舉式的分析方法將要測試約1040種組合,這個天文數字的計算時間不是現實狀況可以負擔的。為了解決這個問題,我們套用了演化上的達爾文天擇學說,藉由基因組合間的多樣性,透過彼此競爭保留表現較佳的組合,再讓這些優勢組合彼此混合變異以找出具有最好預測效果的基因群,此作法即為基因演算法(genetic algorithm)的概念。最終,我們利用機器學習方法中的極度梯度提升(extreme gradient boosting, XGboost)演算法在找出的基因組合上建構預測模型,未來新病患應用時僅需檢測這些基因表現量便可作為其是否復發的生物標記,且這樣的預測模型在獨立的資料集上顯示了良好的敏感度(74-100%),亦即我們可以找出具有復發高風險的卵巢癌病患,採取較為積極的治療方法。
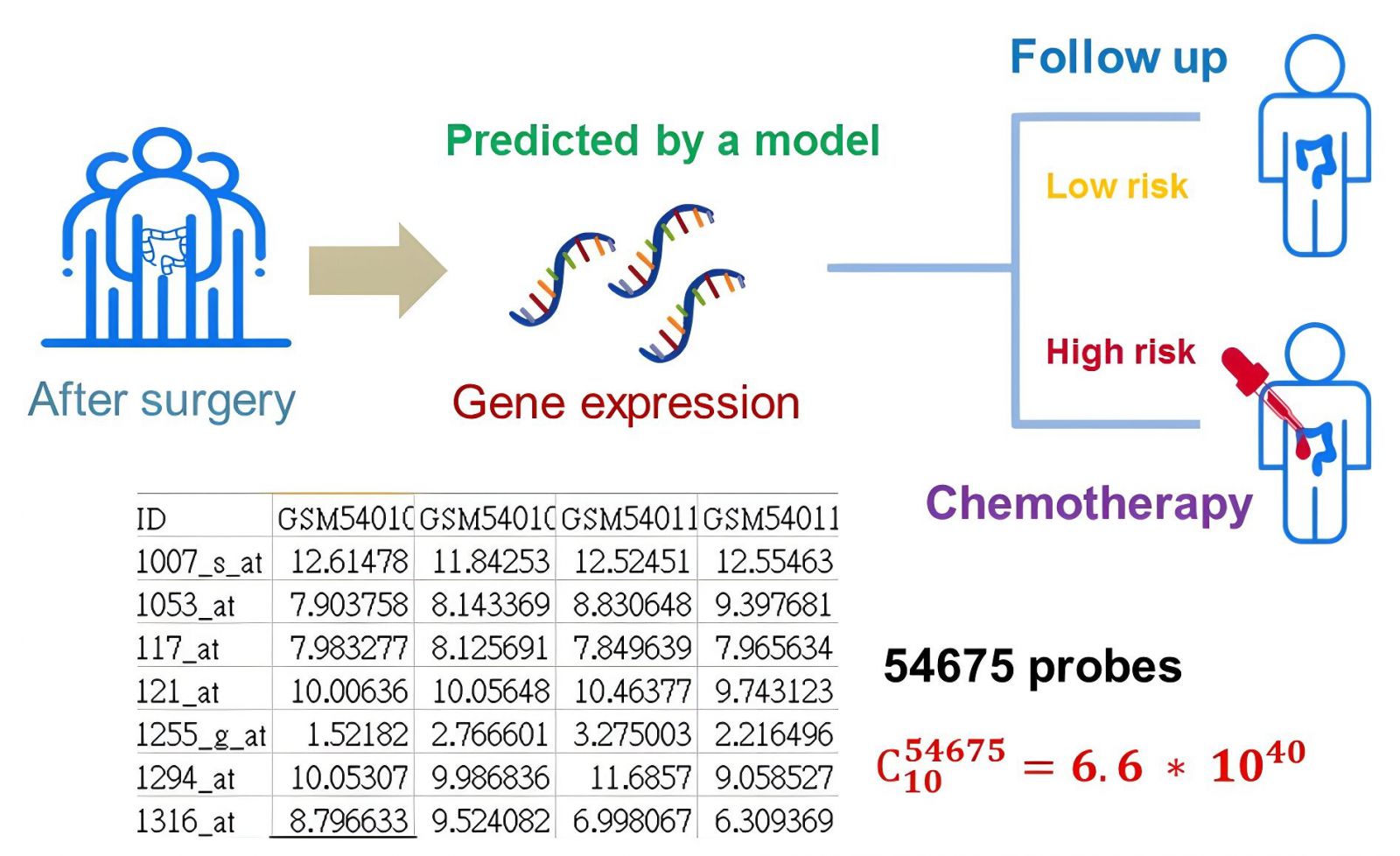
圖2:卵巢癌病患的基因表現量評估復發風險示意圖。
癌症存活預測
除了基因資料的快速累積,由於電腦科技的快速發展,我們已經能夠有完善的系統追蹤且記錄大量疾病病患的生理狀況及疾病特徵,因此,是否可以透過這些數據提供疾病進程更好的預測成為科學界重要的努力方向。舉例來說,當病患被診斷出癌症時,我們想問的問題不出下列幾個:五年存活率有多少?是否要接受某些特定治療?治療的效果如何?過往相似病患的情形如何等等。臺灣在癌症資料收集上於民國68年即開始建立癌症登記系統,而國家級的癌症登記中心則是在民國85年7月開始收集資料,提供了臺灣癌症流行病學研究上的重要基石。在這個優良的基礎上,我們與臺灣的癌登中心開始合作嘗試建構臺灣本土的癌症病患存活預測模型,舉例來說,首先建構完成的系統即為乳癌病患的存活模型(圖3)。使用者僅需在網頁介面上打入病患相關的基本人口學變項以及腫瘤情形,網頁背後的預測模型便可以即時的利用臺灣本土數據回答病患的存活機率,使用者更可以直接調整接受治療的型態來評估治療的效果,我們希望藉由科學數據的提供讓臨床醫師與病患及家屬間的交流能夠更有效率,達到最終醫病共享的目標。


圖3:臺灣癌登資料建構之本土乳癌病患存活預測系統。
結語
整體而言,不論是生物醫學檢測技術或是資訊科技的快速發展,都讓累積健康數據的時間與難度大幅度下降,如何在這些龐大的資料中轉化出重要的數據甚至應用,將會是未來最重要的挑戰(圖4)。生物資訊將扮演極其吃重的角色,從一開始的資料清理,到篩選出重要的生物標記,以及建構出準確的預測模型,都將是生物資訊在健康數據研究中占有一席之地的領域。(本期專題策畫/公衛學院郭柏秀副院長&生農學院李達源副院長)

圖4:未來生物資訊於生物醫學相關研究之整合情形。
參考文獻:
[1]Lu TP, Tsai MH, Lee JM, Hsu CP, Chen PC, Lin CW, et al. Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19(10):2590-7. doi: 10.1158/1055-9965.EPI-10-0332. PubMed PMID: 20802022.
[2]Lu TP, Chuang EY, Chen JJ. Identification of reproducible gene expression signatures in lung adenocarcinoma. BMC bioinformatics. 2013;14:371. doi: 10.1186/1471-2105-14-371. PubMed PMID: 24369726; PubMed Central PMCID: PMC3877965.
[3]Huang CC, Chan SY, Lee WC, Chiang CJ, Lu TP, Cheng SH. Development of a prediction model for breast cancer based on the national cancer registry in Taiwan. Breast cancer research : BCR. 2019;21(1):92. doi: 10.1186/s13058-019-1172-6. PubMed PMID: 31409418; PubMed Central PMCID: PMC6691540.
[4]Hsiao YW, Tao CL, Chuang EY, Lu TP. A risk prediction model of gene signatures in ovarian cancer through bagging of GA-XGBoost models. Journal of Advanced Research. 2020. doi: https://doi.org/10.1016/j.jare.2020.11.006.
盧子彬小檔案
臺大生命科學系及電機系畢業,臺大生醫電子與資訊學研究所博士。現任臺大公共衛生學系教授、公共衛生學會秘書長、臺大醫院外科部兼任研究員。研究興趣為藉由生物資訊方法回答生物醫學問題,並希望可以將學術研究成果轉化為實際可以應用之技術。
